Chapter 6 Genome-Wide Association Study (GWAS)
In {bigstatsr}, you can perform both standard linear and logistic regressions GWAS, using either big_univLinReg() or big_univLogReg().
Function big_univLinReg() should be very fast, while big_univLogReg() is slower.
This type of association, where each variable is considered independently, can be performed for any type of FBM (i.e. it does not have to be a genotype matrix). This is why these two functions are in package {bigstatsr}, and not {bigsnpr}.
6.1 Example
Let us reuse the data prepared in 4.3 and the PCs in 5.2.
#> Loading required package: bigstatsrThe clinical data includes age, sex, high-density lipoprotein (HDL)-cholesterol (hdl), low-density lipoprotein (LDL)-cholesterol (ldl), triglycerides (tg) and coronary artery disease status (CAD).
For the set of covariates, we will use sex, age, and the first 6 PCs:
You probably should not account for other information such as cholesterol as they are heritable covariates (Aschard, Vilhjálmsson, Joshi, Price, & Kraft, 2015; Day, Loh, Scott, Ong, & Perry, 2016).
G <- obj.bigsnp$genotypes
y <- obj.bigsnp$fam$CAD
ind.gwas <- which(!is.na(y) & complete.cases(covar))To only use a subset of the data stored as an FBM (G here), you should almost never make a copy of the data; instead, use parameters ind.row (or ind.train) and ind.col to apply functions to a subset of the data.
Let us perform a case-control GWAS for CAD:
gwas <- runonce::save_run(
big_univLogReg(G, y[ind.gwas], ind.train = ind.gwas,
covar.train = covar[ind.gwas, ], ncores = nb_cores()),
file = "tmp-data/GWAS_CAD.rds")#> user system elapsed
#> 0.13 0.07 136.54This takes about two minutes with 4 cores on my laptop. Note that big_univLinReg() takes two seconds only, and should give very similar p-values, if you just need something quick.
CHR <- obj.bigsnp$map$chromosome
POS <- obj.bigsnp$map$physical.pos
snp_manhattan(gwas, CHR, POS, npoints = 50e3) +
ggplot2::geom_hline(yintercept = -log10(5e-8), linetype = 2, color = "red")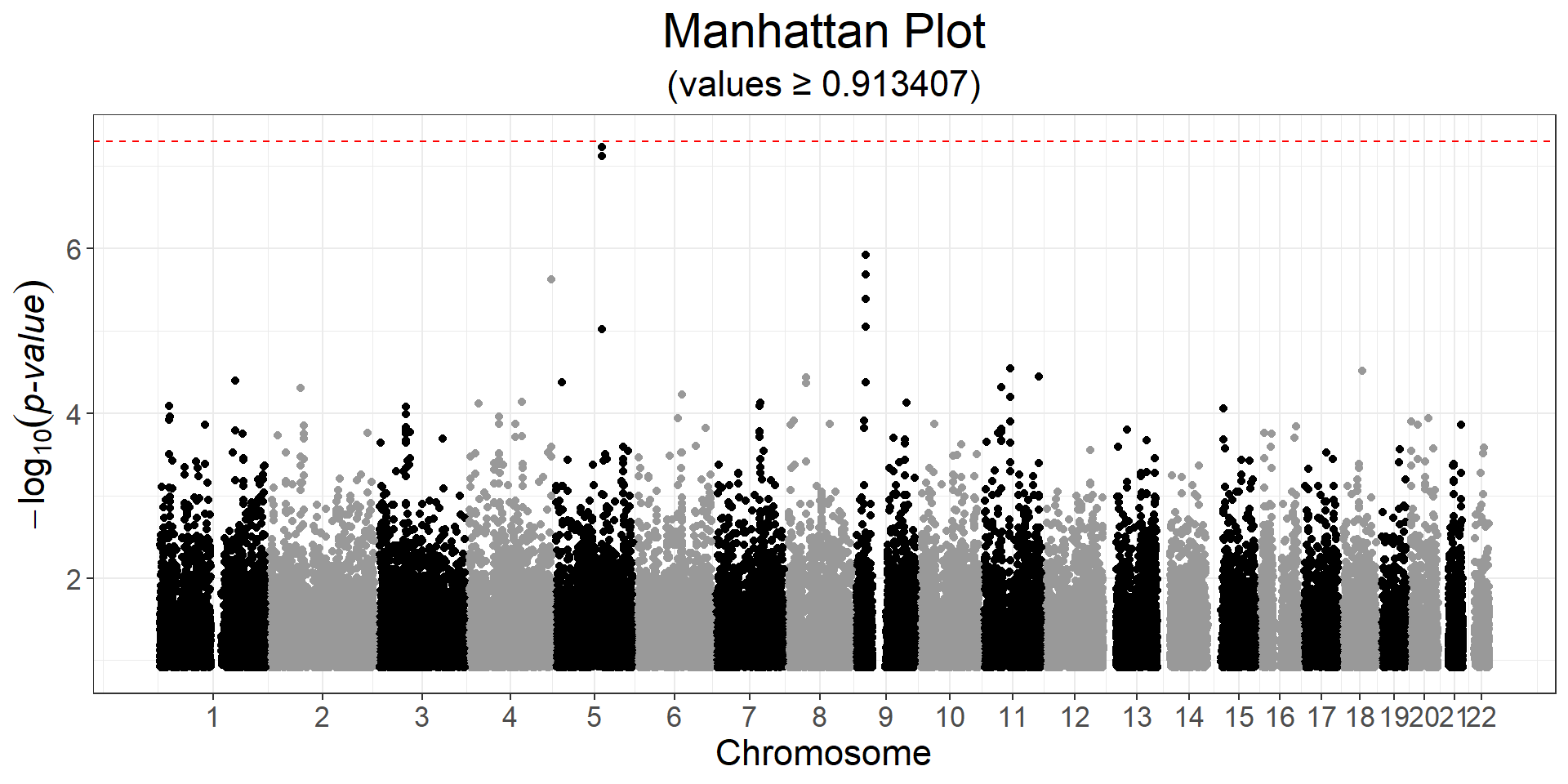 Here, nothing is genome-wide significant because of the small sample size.
Here, nothing is genome-wide significant because of the small sample size.
y2 <- obj.bigsnp$fam$hdl
ind.gwas2 <- which(!is.na(y2) & complete.cases(covar))
gwas2 <- big_univLinReg(G, y2[ind.gwas2], ind.train = ind.gwas2,
covar.train = covar[ind.gwas2, ], ncores = nb_cores())
snp_manhattan(gwas2, CHR, POS, npoints = 50e3) +
ggplot2::geom_hline(yintercept = -log10(5e-8), linetype = 2, color = "red")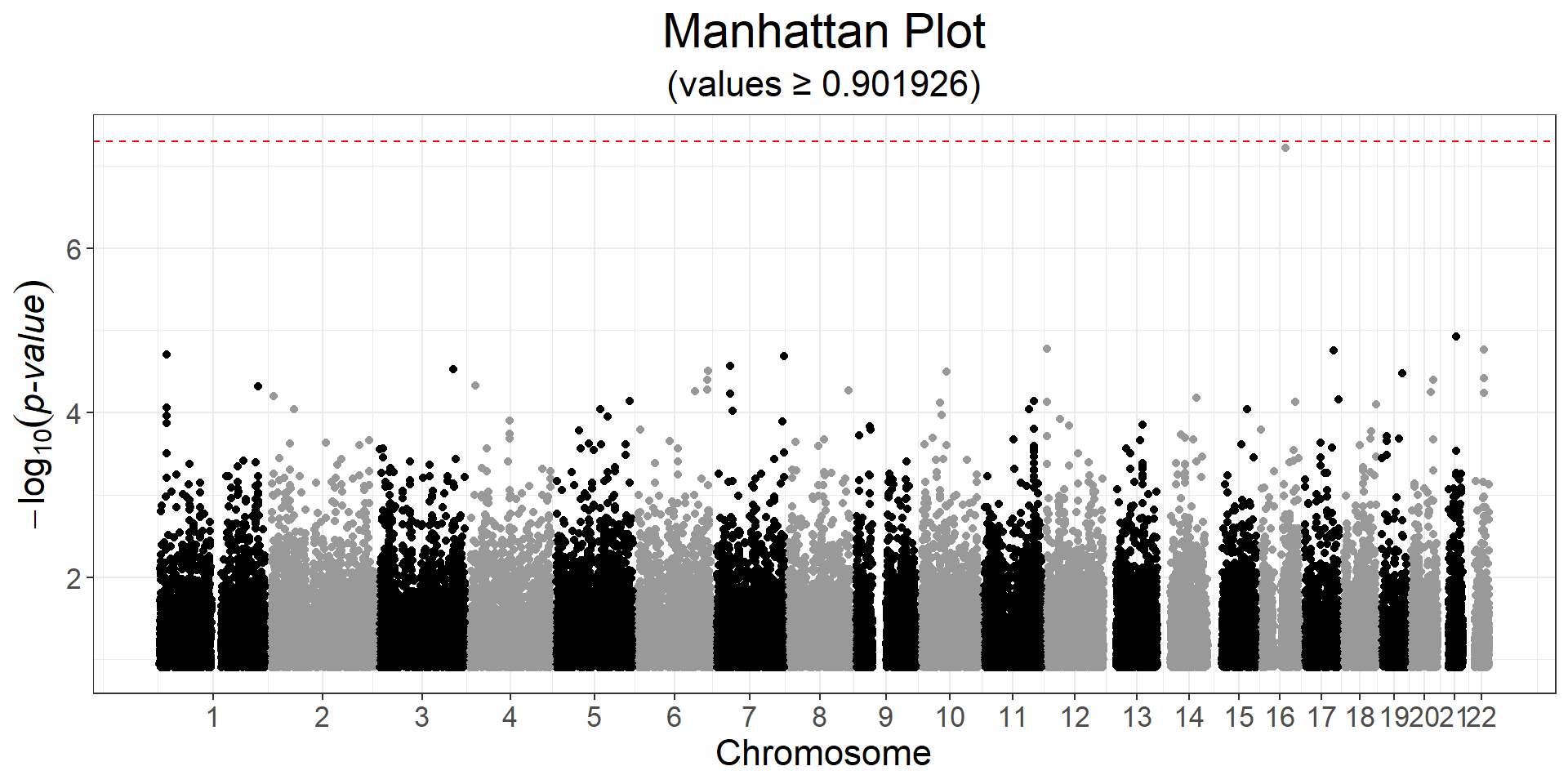
Some other example code:
- GWAS in iPSYCH; you can perform the GWAS on multiple nodes in parallel that would each process a chunk of the variants only
- GWAS for very large data and multiple phenotypes; you should perform the GWAS for all phenotypes for a “small” chunk of columns to avoid repeated access from disk, and can process these chunks on multiple nodes in parallel
- some template for {future.batchtools} when using Slurm