Method to detect genetic markers involved in biological adaptation. This provides a statistical tool for outlier detection based on Principal Component Analysis. This corresponds to the statistic based on mahalanobis distance, as implemented in package pcadapt.
snp_pcadapt(
G,
U.row,
ind.row = rows_along(G),
ind.col = cols_along(G),
ncores = 1
)
bed_pcadapt(
obj.bed,
U.row,
ind.row = rows_along(obj.bed),
ind.col = cols_along(obj.bed),
ncores = 1
)Arguments
- G
A FBM.code256 (typically
<bigSNP>$genotypes).
You shouldn't have missing values. Also, remember to do quality control, e.g. some algorithms in this package won't work if you use SNPs with 0 MAF.- U.row
Left singular vectors (not scores, \(U^T U = I\)) corresponding to
ind.row.- ind.row
An optional vector of the row indices (individuals) that are used. If not specified, all rows are used.
Don't use negative indices.- ind.col
An optional vector of the column indices (SNPs) that are used. If not specified, all columns are used.
Don't use negative indices.- ncores
Number of cores used. Default doesn't use parallelism. You may use
bigstatsr::nb_cores().- obj.bed
Object of type bed, which is the mapping of some bed file. Use
obj.bed <- bed(bedfile)to get this object.
Value
An object of classes mhtest and data.frame returning one
score by SNP. See methods(class = "mhtest").
References
Luu, K., Bazin, E., & Blum, M. G. (2017). pcadapt: an R package to perform genome scans for selection based on principal component analysis. Molecular ecology resources, 17(1), 67-77.
See also
snp_manhattan, snp_qq and snp_gc.
Examples
test <- snp_attachExtdata()
G <- test$genotypes
obj.svd <- big_SVD(G, fun.scaling = snp_scaleBinom(), k = 10)
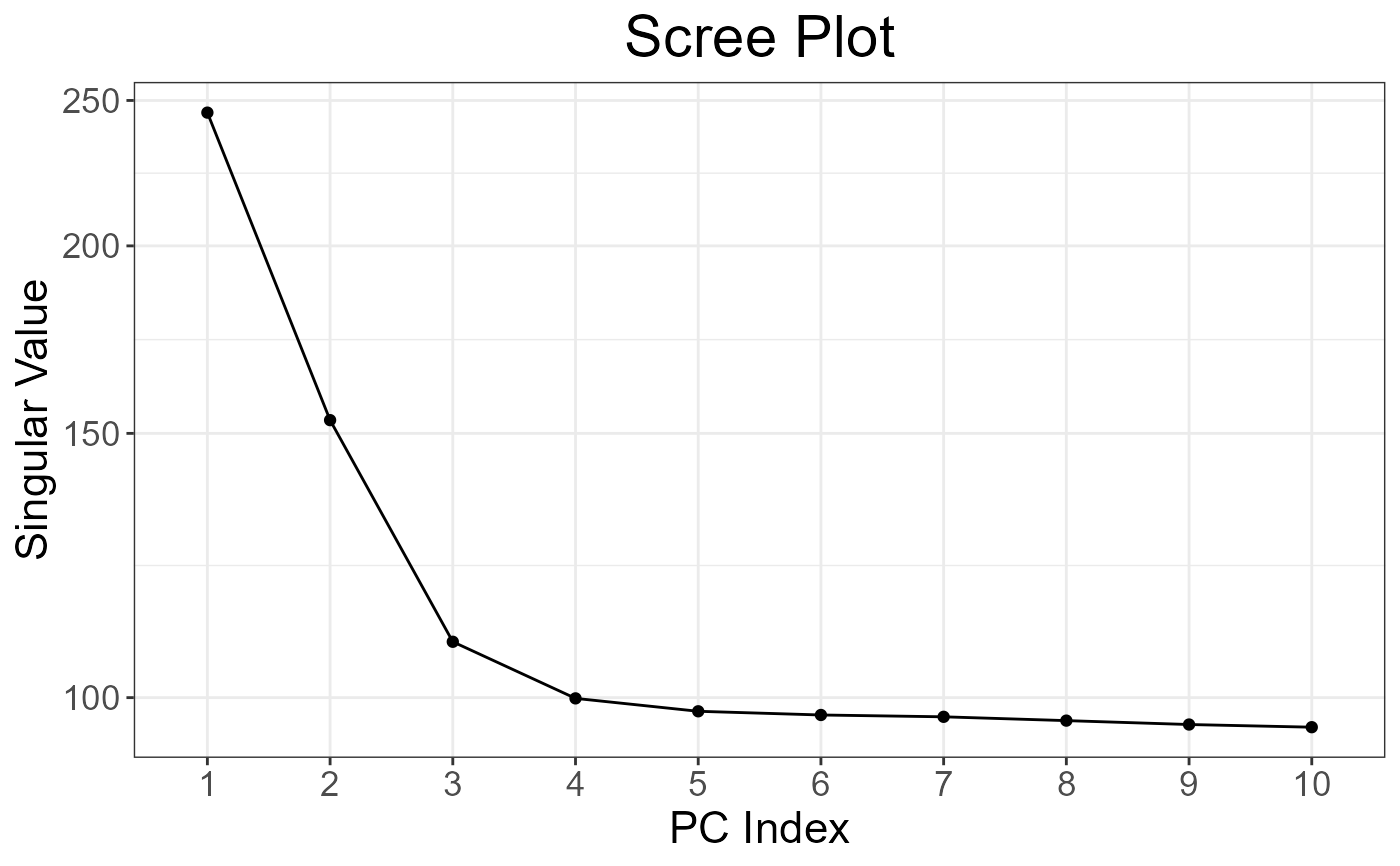
plot(obj.svd) # there seems to be 3 "significant" components
 pcadapt <- snp_pcadapt(G, obj.svd$u[, 1:3])
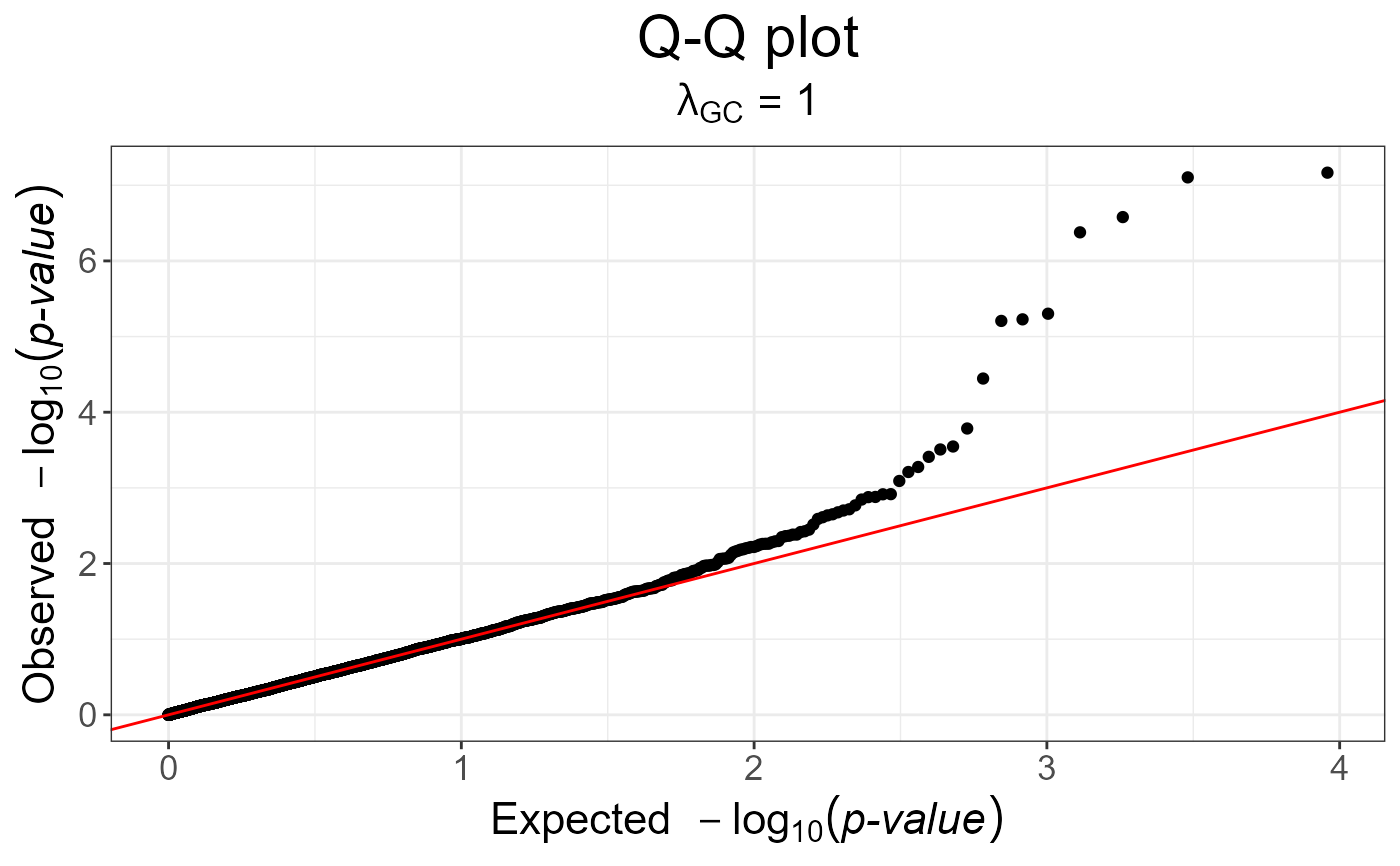
snp_qq(pcadapt)
pcadapt <- snp_pcadapt(G, obj.svd$u[, 1:3])
snp_qq(pcadapt)